研究兴趣
研究概要
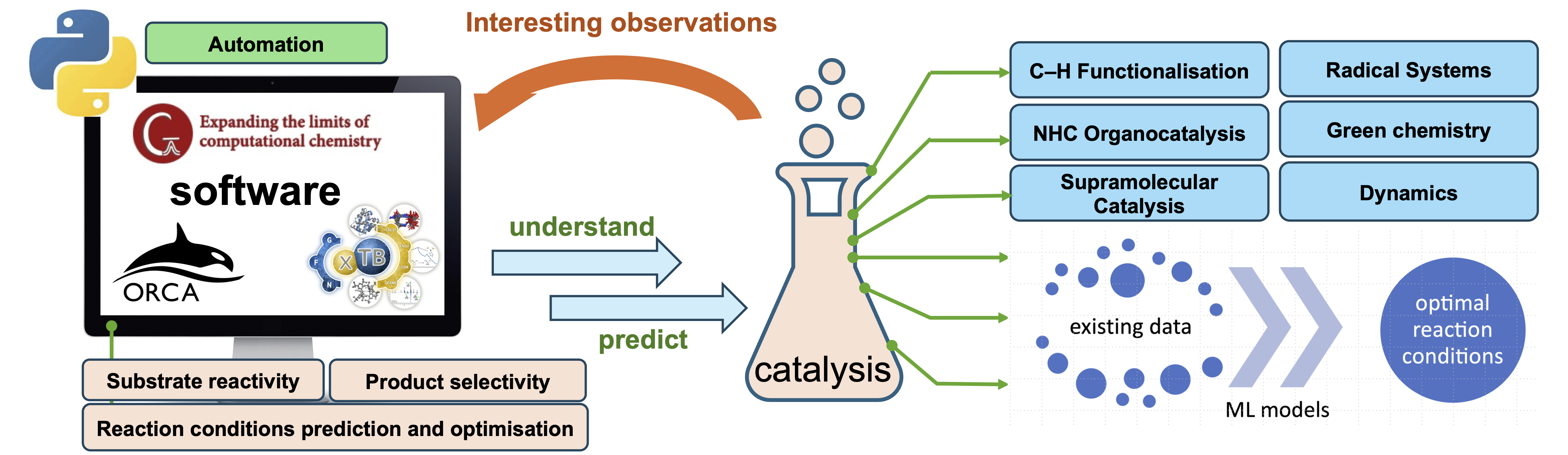
我们致力于运用电子结构理论计算,主要依赖于 密度泛函理论 (DFT),对具有学术和工业意义的复杂有机和有机金属催化体系进行计算机理研究。我们目前专注于过渡金属催化的C–H官能化以及用于复杂分子构建的有机催化不对称合成。我们正在开发应用机器学习原子间势 (MLIPs) 的能力,以研究传统上难以用静态DFT准确模拟的化学体系,例如动态和熵效应。我们的兴趣还包括编码和工作流程开发,以促进高通量量子化学研究和机器学习应用,以达到优化计算研究。
1.计算均相催化
1.1 有机金属催化
C–H官能化
过渡金属 (TM)
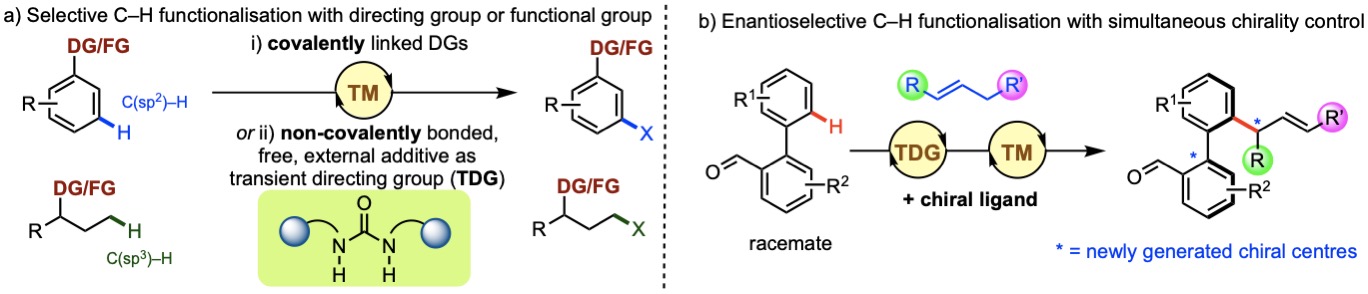
催化的C–H官能化为有机合成提供了许多机会,通过从许多有机分子中存在的未反应、常见的C–H键中形成有用的C–X键。使用TM来选择性官能化C–H键尤其具有吸引力,因为它提供了原子经济的方法,可以将小烷烃转化为更高价值的官能化分子,或直接操作具有其他官能团的复杂分子。此类TM催化的C–H官能化通常依赖于导向基团
(DGs)
来实现目标C–H活化。无导向基团的方法开发避免了引入和随后移除共价DGs,从而提高步骤经济性。我们对研究此类DG辅助和DG-free催化体系的机制感兴趣。
a)
使用共价/非共价导向基团或底物中固有官能团的过渡金属催化选择性C–H活化。b)
同时控制手性的对映选择性C–H官能化。
Pd(OAc)2催化的炔基化反应的吉布斯能量分布图。MPAA配体降低了C–H活化的障碍(ts-1'优于ts-1)。此外,醋酸银有助于降低β-溴化物消除的障碍(ts-4'优于ts-4)。
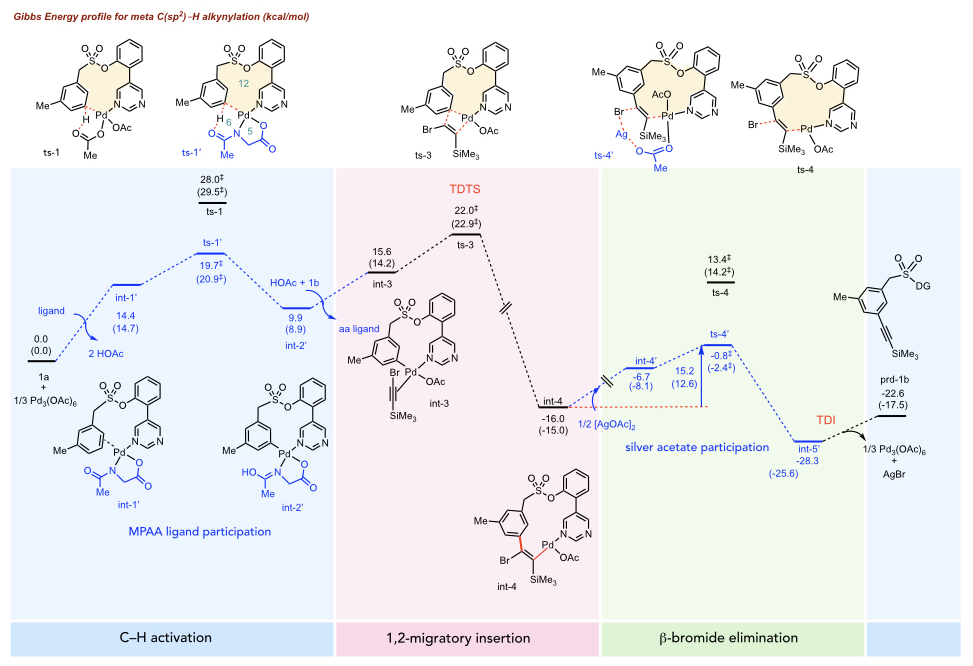
在研究反应机制时,我们通常需要理解导致不同化学选择性的分子起源。我们可能还需要比较不同底物的化学反应性。例如,在芳烃的meta-选择性C–H官能化中(上图),我们能够通过计算阐明meta-相对于para-或ortho-选择性的独特起源。我们通过比较每个位点的周转频率决定过渡态
(TDTSs)
来实现这一点(中图)。我们能够通过等键反应焓计算来比较不同大小的钯环中间体的环张力,从而计算不同过渡态中环张力的差异。
高度meta-选择性的芳烃烯丙基化(第一图)、芳烃位点选择性的相对障碍(中图)以及E-烯丙基产物形成相对于Z-烯丙基和苯乙烯基产物形成的环张力(最后图)。
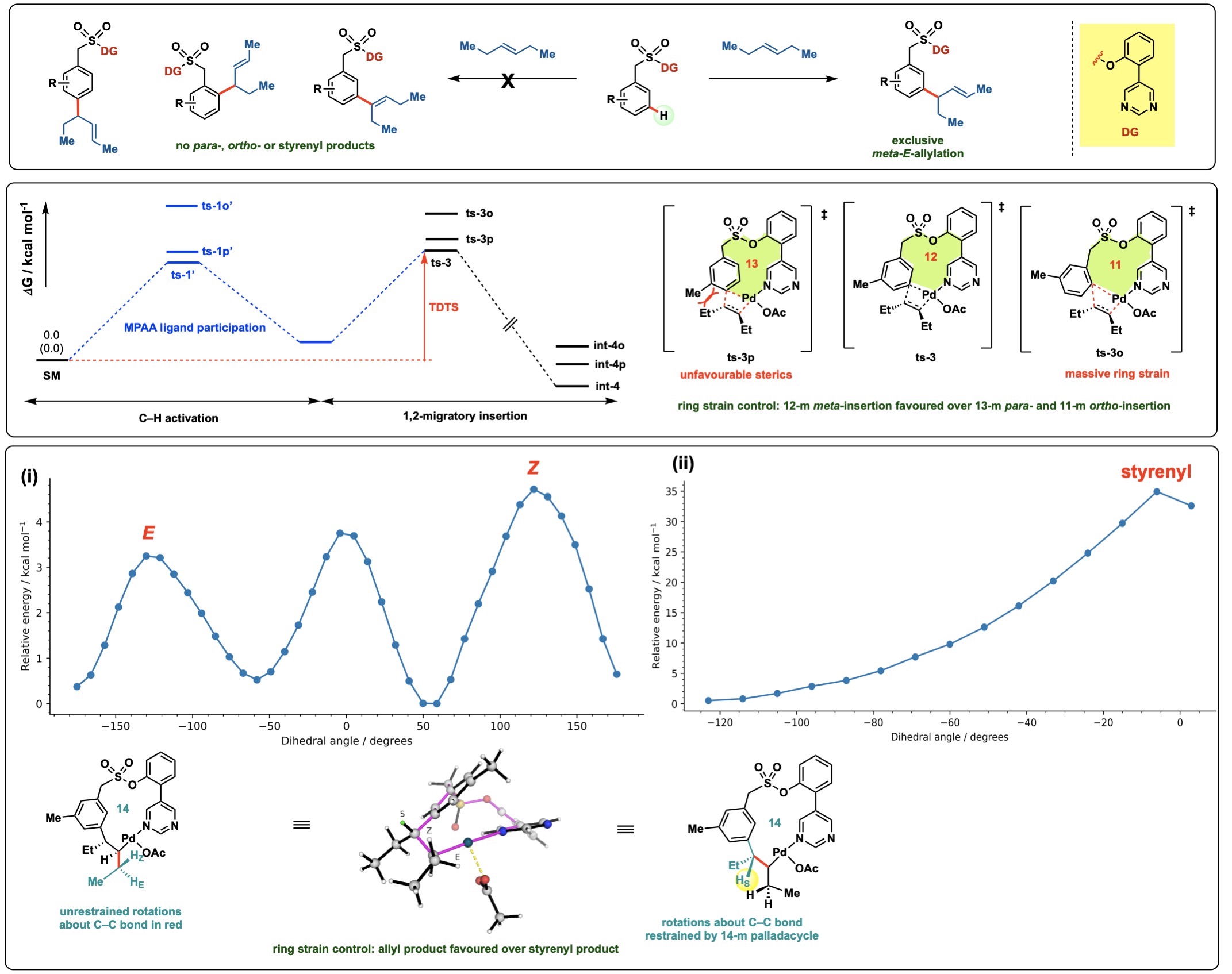
钯催化剂:
1. Angew. Chem. Int. Ed., 2019, 58, 5633–5638.
2. Angew. Chem. Int. Ed., 2019, 58, 10353–10360.
3. J. Am. Chem. Soc., 2020, 142, 8, 3762–3774.
4. J. Am. Chem. Soc. 2022, 144, 27, 12032–12042.
5. Nat. Chem., 2023, 15, 1626–1635.
6. ACS Catal., 2023, 13, 21, 14000–14011.
Rhodium catalysts:
铑催化剂:
1. Chem. Sci., 2023, 14, 11381–11388.
C–C键偶联
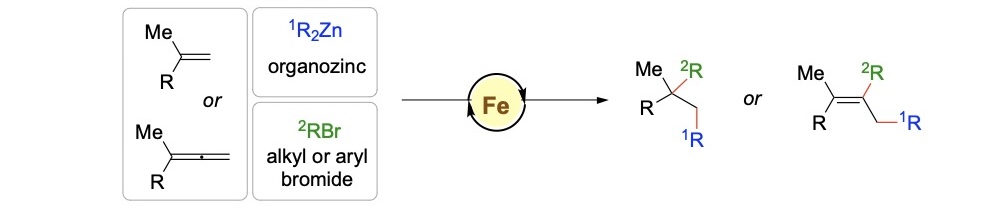
形成C–C键的能力是有机化合物合成的核心,包括药物、农药和天然产物,使化学家能够从简单的起始材料构建复杂分子。控制分子上键形成的位置(区域选择性)至关重要,特别是对于具有多个反应位点的分子。结合这些挑战,理想的催化剂需要廉价且可持续。与实验学家合作,我们专注于阐明铁催化的交叉偶联反应的机制(上图),该反应将多个组分聚在一起,在铁催化下形成复杂分子。由于铁的可变自旋态(高自旋与低自旋)以及铁可能涉及不同氧化态(Fe(I)、Fe(II)、Fe(III)),仅使用实验技术可能不足以完全理解催化循环。我们使用DFT计算来理解此类复杂转化的机制(下图)。
内球面自由基-自由基C–C偶联的内禀反应坐标 (IRC) 动画。
外球面还原消除的内禀反应坐标 (IRC) 动画。
上图:铁催化的多组分连结C–C交叉偶联反应。下图:铁催化连结烷基化的计算吉布斯能量分布图。DFT:SMD(THF)-UB3LYP-D3BJ(UMN15)/def2-TZVP//UB3LYP-D3BJ/def2-SVP。能量值前的上标表示各个化合物的自旋态。
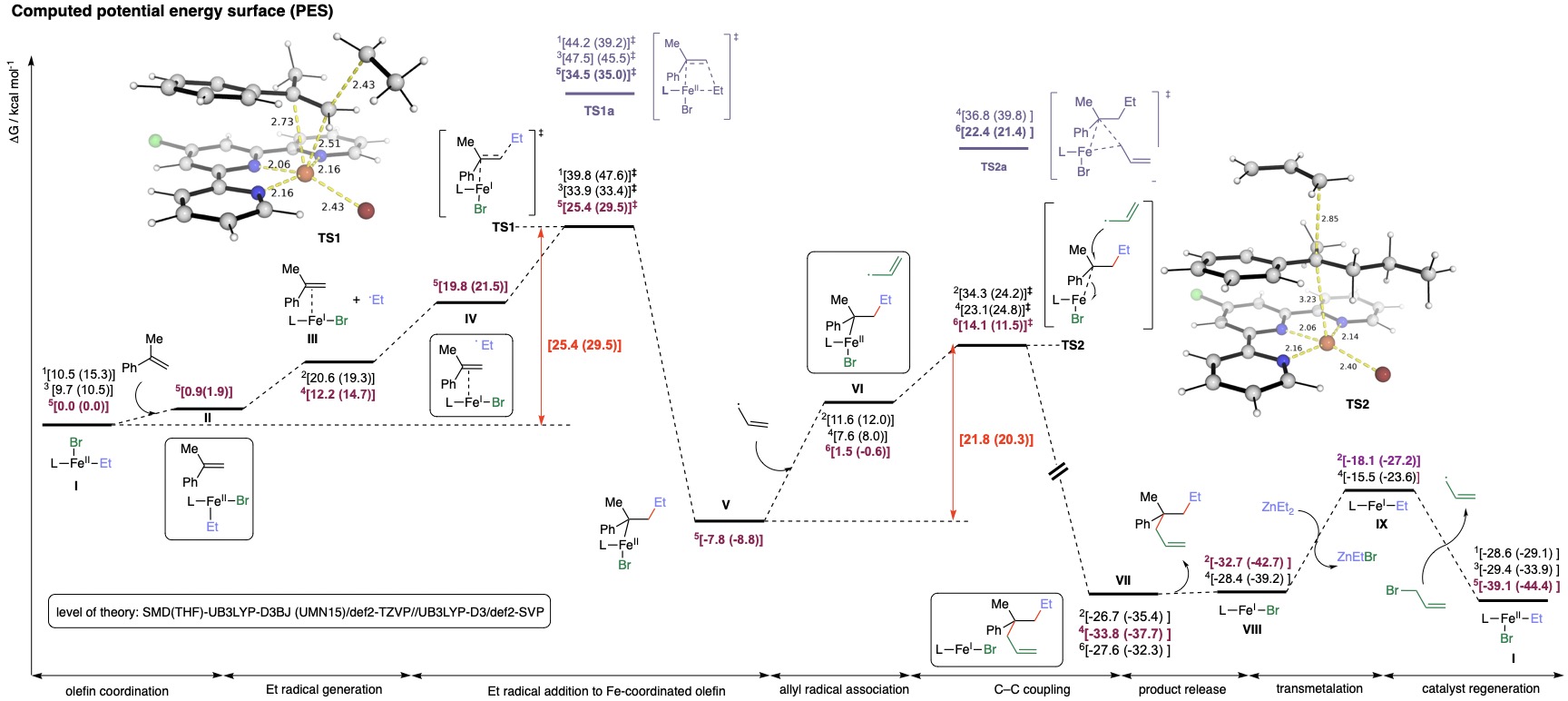
铁催化剂:
1. Nat. Catal., 2024, 7, 321–329.
2. Nat. Synth., 2025, 4, 116–123.
1.2 有机催化与不对称催化
N-杂环卡宾 (NHC) 催化
NHC有机催化剂提供多样的催化活化模式:NHC活化的物种可以通过羰基碳的逆极加成与亲电试剂反应,或通过氧化后的Brewslow中间体与亲核试剂反应。这为计算工具研究详细机制提供了许多途径。我们实验室将研究无金属有机催化,包括卡宾催化。我们旨在为手性分子构建的有机催化领域做出贡献,目标是发现新的活化模式并官能化难以反应的惰性分子,如羰基化合物、酮烯和醇类,用于药物、农药和特种化学品合成的应用。我们将被探索一些领域挑战,如如何将NHC用作不对称Brønsted碱。
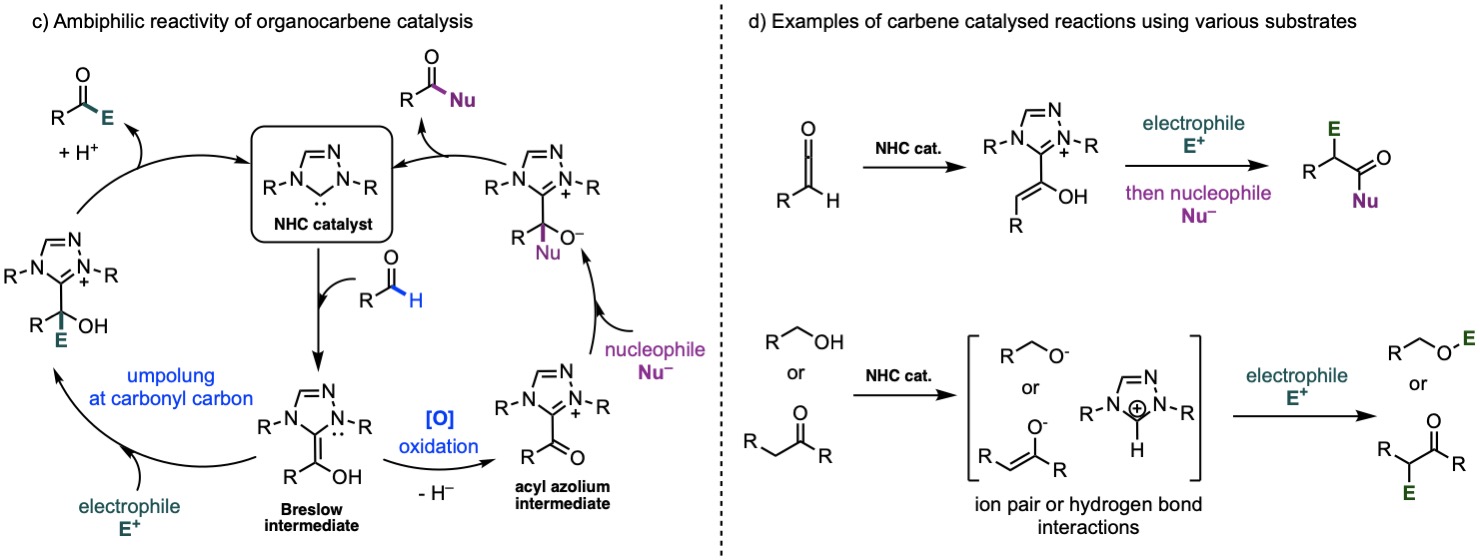
c) 有机卡宾催化剂的两亲反应性。d) 使用各种底物的卡宾催化反应示例。
手性NHC卡宾已被用作有机催化剂,通过酰基唑鎓中间体实现立体和对映诱导。由于手性卡宾赋予的手性,亲核试剂可能选择性地从羰基团的Re-面或Si-面进攻(例如,单糖的羟基进攻)。
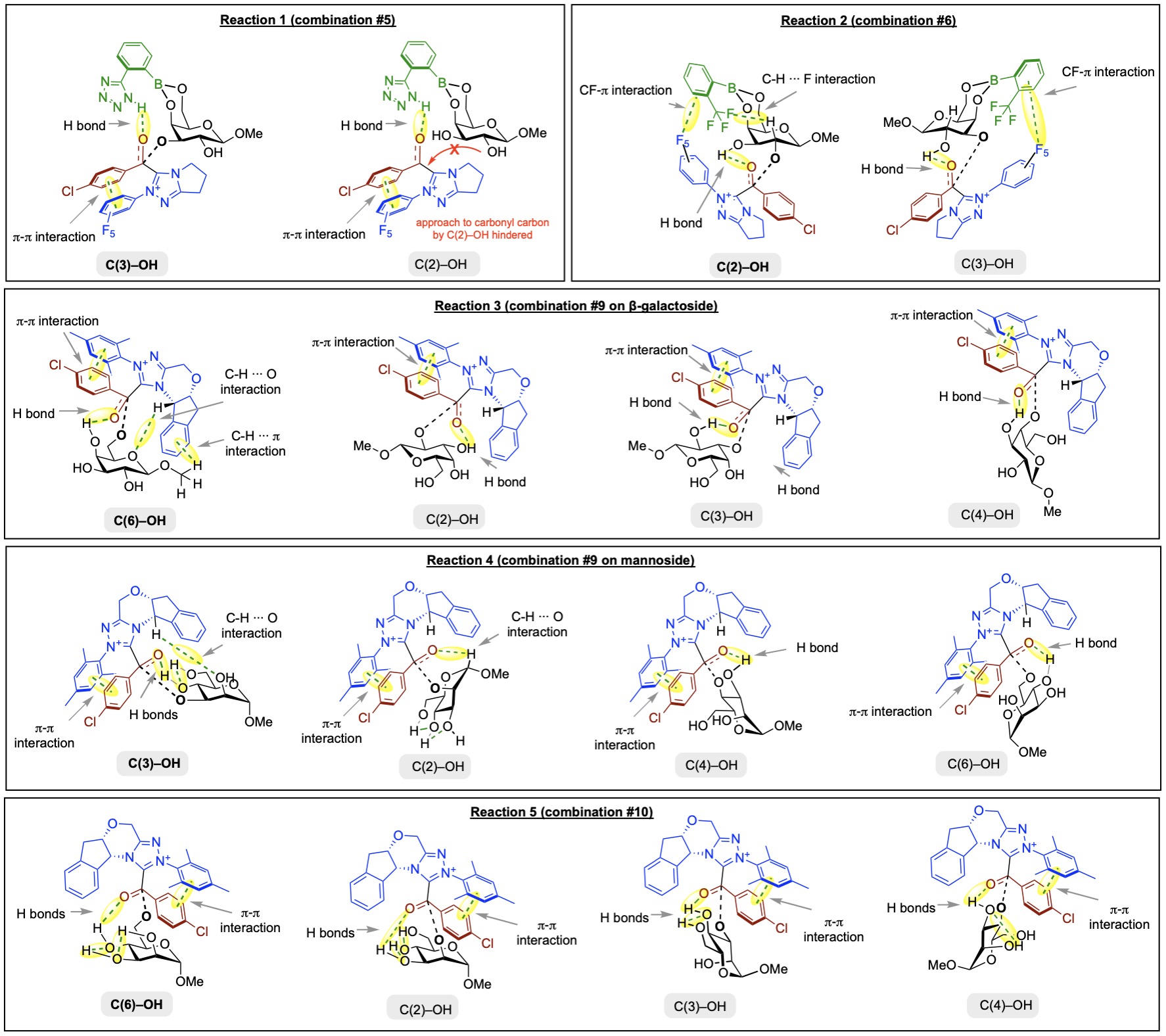
对单糖OH基团区域选择性酰化的模型反应的计算研究。探索了不同硼酸和卡宾组合对不同立体化学单糖的区域选择性控制。
手性卡宾催化剂还可能参与形成具有偏向立体和电子效应的手性中间体,使其能够对映选择性地进攻前手性底物的一面而不是另一面。例如,在下述反应中,手性卡宾形成氮杂-Breslow中间体,可被氧化为三氮二烯中间体,进攻异吲哚的Si-面而非Re-面。
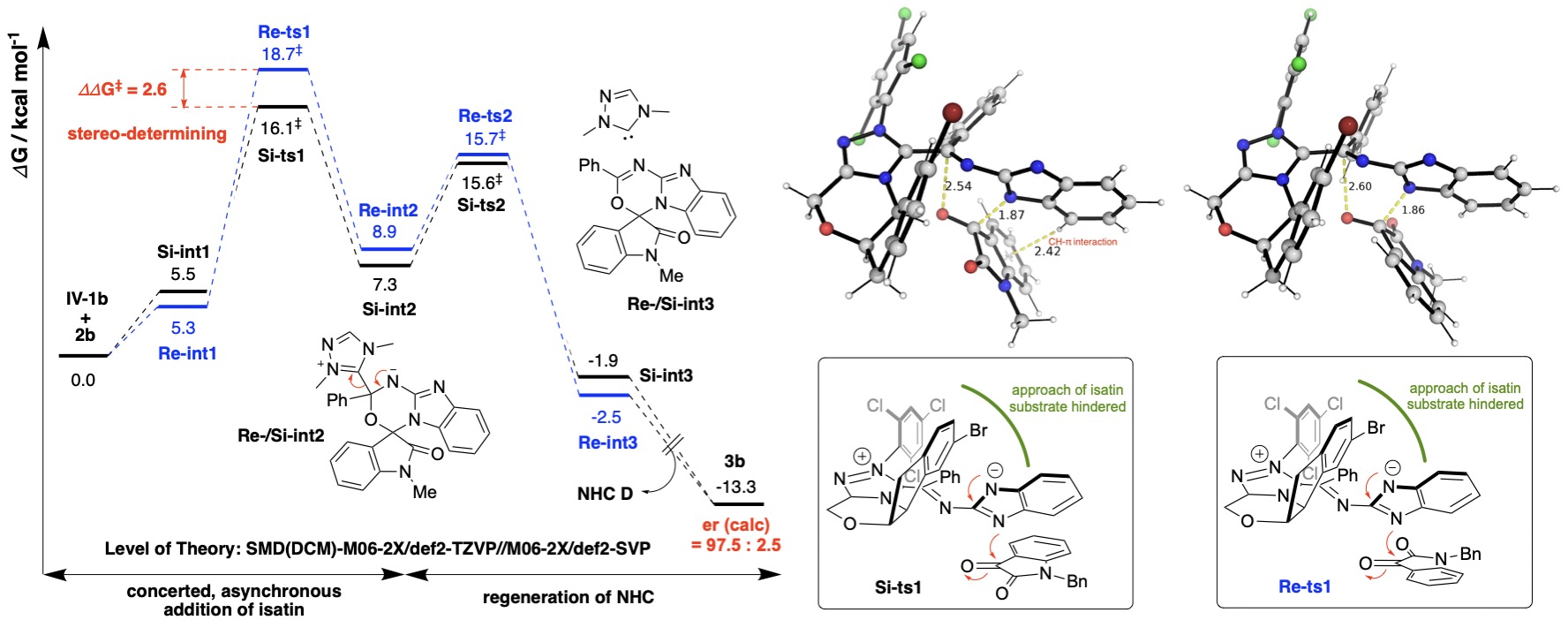
卡宾形成中间体对异吲哚的对映选择性进攻的吉布斯能量分布图及过渡态结构。
为准确预测选择性步骤,我们需要进行详细的构象采样(并对所有竞争途径的TS构象进行Boltzmann Weighting,如果构象能量非常接近的话),因为错误使用构象可能导致错误预测,如下图所示。我们通常对定位的TS结构进行CREST构象采样,约束关键反应键,并根据系统大小使用5-20个最低xTB能量结构进行DFT精炼的进一步TS优化。
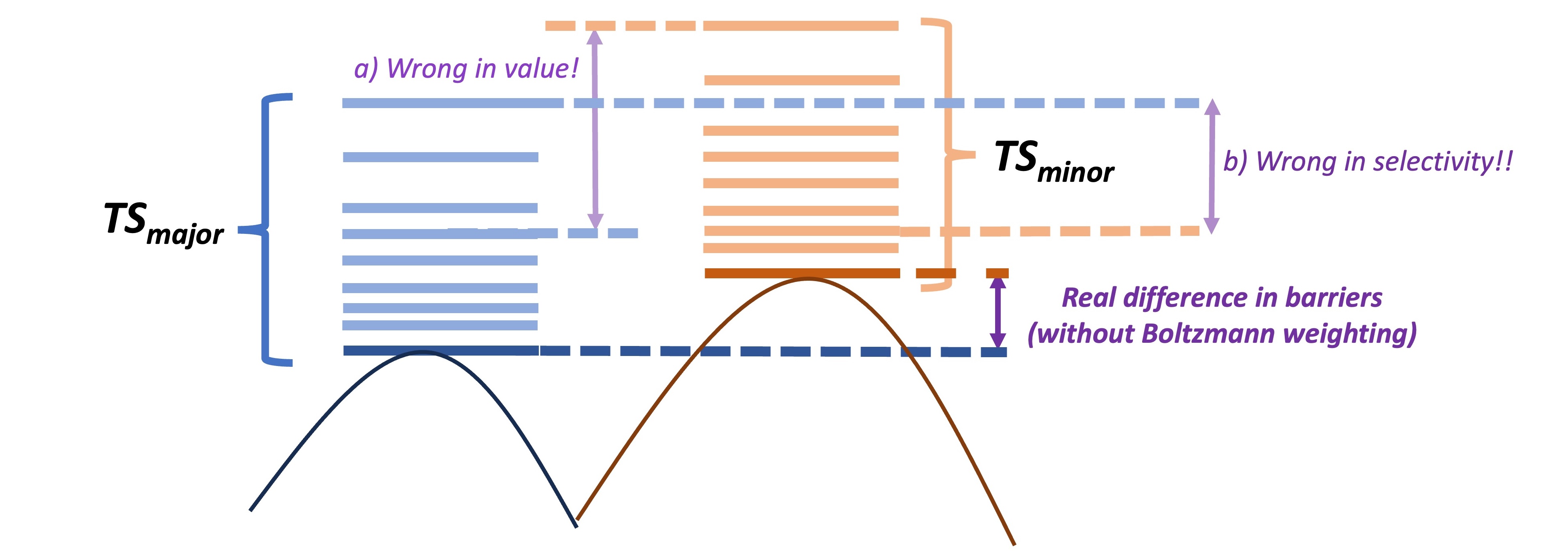
构象采样对准确预测选择性的挑战。使用非最低能量TS结构可能导致错误的选择性比率预测(a)甚至相反的选择性预测(b)!
有机卡宾:
1. Angew. Chem. Int. Ed., 2021, 60, 7906–7912.
2. Org. Chem. Front., 2021, 8, 2413–2419.
3. Nat. Commun., 2022, 13, 36.
4. J. Am. Chem. Soc., 2022, 144, 12, 5441–5449.
5. Chem, 2022, 8, 1518–1534.
6. Angew. Chem. Int. Ed. 2023, 62, e202211977.
7. Angew. Chem. Int. Ed., 2024, 63, e202404979.
8. J. Am. Chem. Soc., 2024, 146, 36, 25350–25360.
Chiral phosphoric acid:
1. Angew. Chem. Int. Ed. 2023, 62, e202306864.
2. 化学的机器学习
传统催化体系的DFT研究通常依赖于带有隐式溶剂化模型的静态DFT。虽然这种方法在较低计算成本下提供了解释和预测成功,但当溶剂分子积极参与反应且需要显式溶剂化时可能不足。然而,在DFT水平上研究具有显式溶剂化的催化体系在计算上可能需要很多计算力和计算资源,特别是对于需要大量轨迹的大型动态系统进行模拟。
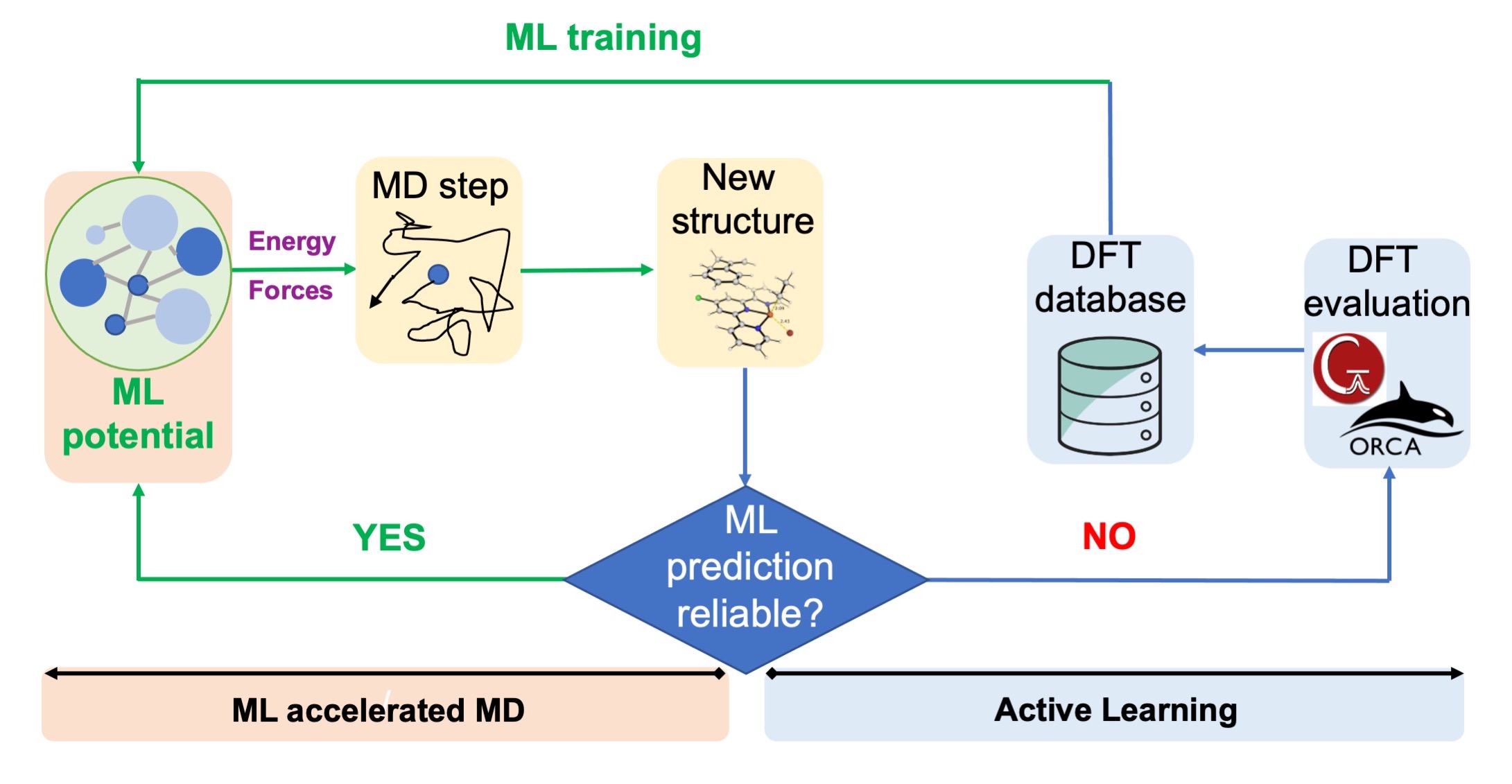
利用主动学习策略加速的机器学习分子动力学 (MD) 模拟。图片改编自 Chem. Sci.,2023,14,8338
在我们关于异相催化中反应化学的MLIPs工作的基础上,我们目前正在探索如何将此类MLIPs应用于研究均相催化体系,其中显式溶剂和大熵变起重要作用。我们希望利用主动学习来降低训练成本,而无需担心MLIP势的迁移性。有了这些工具,研究昂贵且具有挑战性的系统,如显式溶剂化和大长度及时间尺度上的动态效应,可以在可负担的成本内以DFT级别的精度常规进行。